cdc <- data.frame(
Year = c(1922L,
1923L,1924L,1925L,1926L,1927L,1928L,
1929L,1930L,1931L,1932L,1933L,1934L,1935L,
1936L,1937L,1938L,1939L,1940L,1941L,
1942L,1943L,1944L,1945L,1946L,1947L,1948L,
1949L,1950L,1951L,1952L,1953L,1954L,
1955L,1956L,1957L,1958L,1959L,1960L,
1961L,1962L,1963L,1964L,1965L,1966L,1967L,
1968L,1969L,1970L,1971L,1972L,1973L,
1974L,1975L,1976L,1977L,1978L,1979L,1980L,
1981L,1982L,1983L,1984L,1985L,1986L,
1987L,1988L,1989L,1990L,1991L,1992L,1993L,
1994L,1995L,1996L,1997L,1998L,1999L,
2000L,2001L,2002L,2003L,2004L,2005L,
2006L,2007L,2008L,2009L,2010L,2011L,2012L,
2013L,2014L,2015L,2016L,2017L,2018L,
2019L,2020L,2021L,2022L,2023L,2024L,2025L),
No..Reported.Pertussis.Cases = c(107473,
164191,165418,152003,202210,181411,
161799,197371,166914,172559,215343,179135,
265269,180518,147237,214652,227319,103188,
183866,222202,191383,191890,109873,
133792,109860,156517,74715,69479,120718,
68687,45030,37129,60886,62786,31732,28295,
32148,40005,14809,11468,17749,17135,
13005,6799,7717,9718,4810,3285,4249,
3036,3287,1759,2402,1738,1010,2177,2063,
1623,1730,1248,1895,2463,2276,3589,
4195,2823,3450,4157,4570,2719,4083,6586,
4617,5137,7796,6564,7405,7298,7867,
7580,9771,11647,25827,25616,15632,10454,
13278,16858,27550,18719,48277,28639,
32971,20762,17972,18975,15609,18617,6124,
2116,3044,7063,22538,21996)
)CLass 18: Pertussis Mini Project
Background
Pertussis is a common lung infection caused by the bacteria B.Pertussis
This can infect all ages but is most severe for those under 1 year of age.
The CDC track the number of reported cases in US We can “scrape” those data with the datapasta package.
Q. Make a plot of
year vs ``cases
library(ggplot2)
ggplot(cdc) +
aes(Year, No..Reported.Pertussis.Cases) +
geom_point() +
geom_line()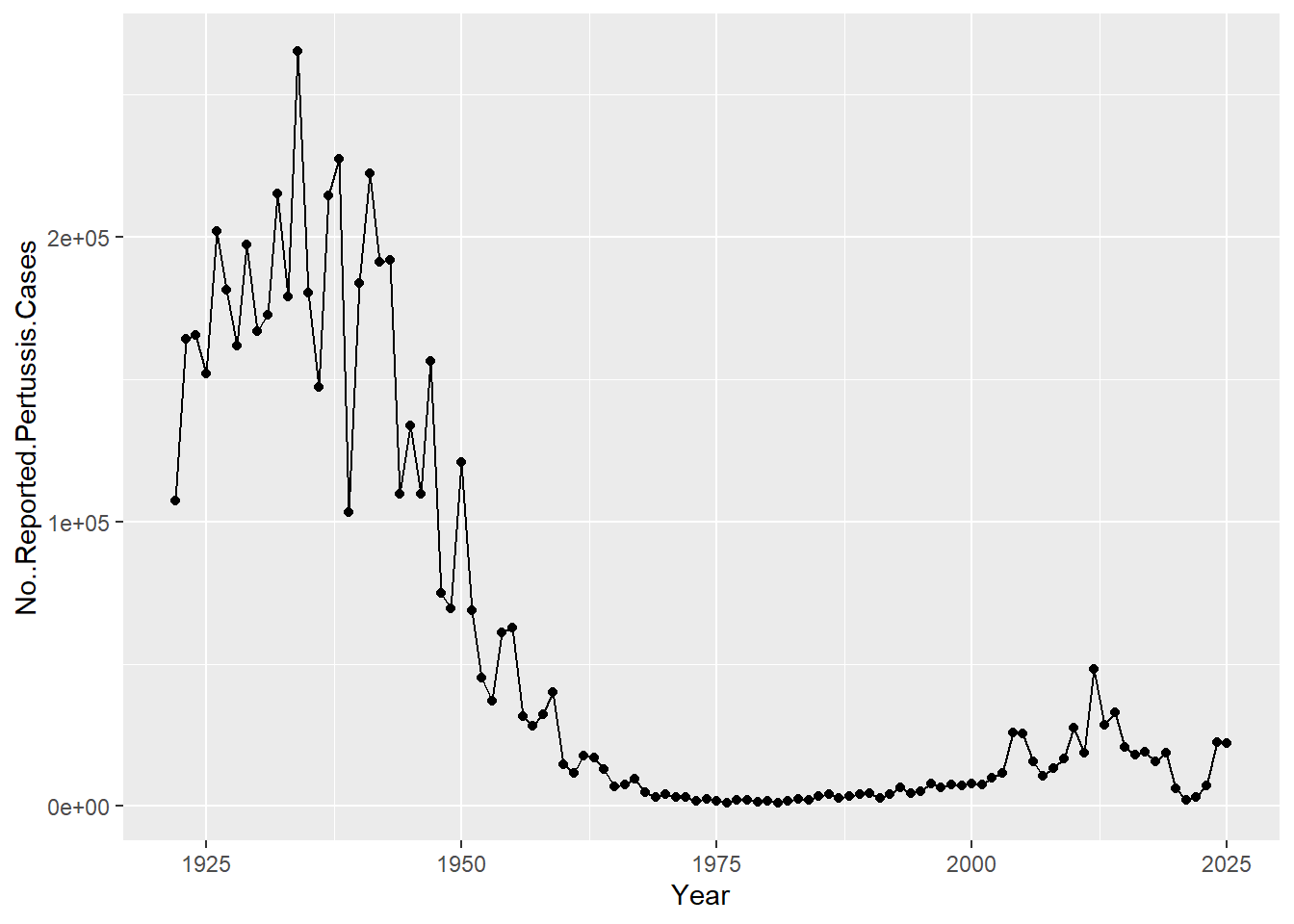
Q. Add some major milestones including the first wP vaccine roll-out (1946), the switch to the newer aP vaccine (1996), the COVID years (2020).
library(ggplot2)
ggplot(cdc) +
aes(Year, No..Reported.Pertussis.Cases) +
geom_point() +
geom_line() +
geom_vline(xintercept= 1946, col="blue", lty=2) +
geom_vline(xintercept= 1996, col="red", lty=2) +
geom_vline(xintercept= 2020, col="gray", lty=2)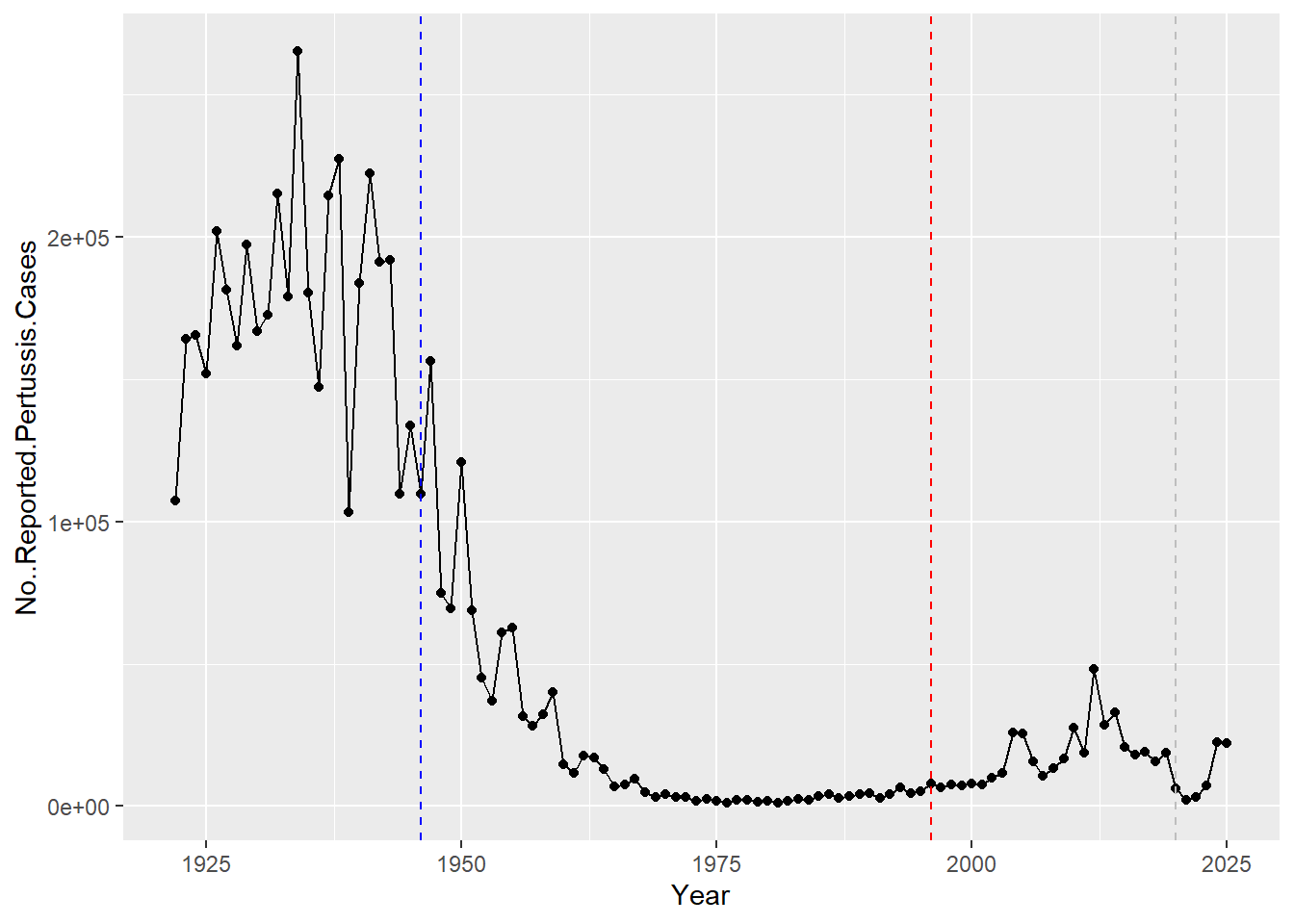
After the switch to the acellular pertussis (aP) vaccine, pertussis cases began rising again, with major peaks such as the 2012 outbreak. coud be due to bacterial evolution and immunity of the vaccine.
Why is this vaccine-preventable disease on the upswing? To answer this question we need to investigate the mechanisms underlying waning protection against pertussis. This requires evaluation of pertussis-specific immune responses over time in wP and aP vaccinated individuals.
CMI-PB project
Computational Models of Immunity - Pertussis Boost project aims to provide the scientific community with this very information.
They make their data abailablke via JSON format returning API. We can read this in R with the read_json() function from the jsonlite package:
library(jsonlite)
subject <- read_json("http://cmi-pb.org/api/v5_1/subject", simplifyVector = TRUE)
head(subject) subject_id infancy_vac biological_sex ethnicity race
1 1 wP Female Not Hispanic or Latino White
2 2 wP Female Not Hispanic or Latino White
3 3 wP Female Unknown White
4 4 wP Male Not Hispanic or Latino Asian
5 5 wP Male Not Hispanic or Latino Asian
6 6 wP Female Not Hispanic or Latino White
year_of_birth date_of_boost dataset
1 1986-01-01 2016-09-12 2020_dataset
2 1968-01-01 2019-01-28 2020_dataset
3 1983-01-01 2016-10-10 2020_dataset
4 1988-01-01 2016-08-29 2020_dataset
5 1991-01-01 2016-08-29 2020_dataset
6 1988-01-01 2016-10-10 2020_datasetQ. How many “wP” and “aP” individuals are in the
subjecttable?
table(subject$infancy_vac)
aP wP
87 85 Q. What is the biological sex breakdown?
table(subject$biological_sex)
Female Male
112 60 Q. In terms of race and gender is this dataset representative of the US population?
table(subject$race, subject$biological_sex)
Female Male
American Indian/Alaska Native 0 1
Asian 32 12
Black or African American 2 3
More Than One Race 15 4
Native Hawaiian or Other Pacific Islander 1 1
Unknown or Not Reported 14 7
White 48 32Let’s read some more database tables:
specimen <- read_json( "http://cmi-pb.org/api/v5_1/specimen", simplifyVector = TRUE)
ab_titer <- read_json( "http://cmi-pb.org/api/v5_1/plasma_ab_titer", simplifyVector = TRUE)head(specimen) specimen_id subject_id actual_day_relative_to_boost
1 1 1 -3
2 2 1 1
3 3 1 3
4 4 1 7
5 5 1 11
6 6 1 32
planned_day_relative_to_boost specimen_type visit
1 0 Blood 1
2 1 Blood 2
3 3 Blood 3
4 7 Blood 4
5 14 Blood 5
6 30 Blood 6head(ab_titer) specimen_id isotype is_antigen_specific antigen MFI MFI_normalised
1 1 IgE FALSE Total 1110.21154 2.493425
2 1 IgE FALSE Total 2708.91616 2.493425
3 1 IgG TRUE PT 68.56614 3.736992
4 1 IgG TRUE PRN 332.12718 2.602350
5 1 IgG TRUE FHA 1887.12263 34.050956
6 1 IgE TRUE ACT 0.10000 1.000000
unit lower_limit_of_detection
1 UG/ML 2.096133
2 IU/ML 29.170000
3 IU/ML 0.530000
4 IU/ML 6.205949
5 IU/ML 4.679535
6 IU/ML 2.816431To analyze this data we need to first “join” (merge/link) the different tables so we have all the data in one place not spread across different tables.
We can use the *_join() family of functions from dplyr to do this
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionmeta <- inner_join(subject, specimen)Joining with `by = join_by(subject_id)`head(meta) subject_id infancy_vac biological_sex ethnicity race
1 1 wP Female Not Hispanic or Latino White
2 1 wP Female Not Hispanic or Latino White
3 1 wP Female Not Hispanic or Latino White
4 1 wP Female Not Hispanic or Latino White
5 1 wP Female Not Hispanic or Latino White
6 1 wP Female Not Hispanic or Latino White
year_of_birth date_of_boost dataset specimen_id
1 1986-01-01 2016-09-12 2020_dataset 1
2 1986-01-01 2016-09-12 2020_dataset 2
3 1986-01-01 2016-09-12 2020_dataset 3
4 1986-01-01 2016-09-12 2020_dataset 4
5 1986-01-01 2016-09-12 2020_dataset 5
6 1986-01-01 2016-09-12 2020_dataset 6
actual_day_relative_to_boost planned_day_relative_to_boost specimen_type
1 -3 0 Blood
2 1 1 Blood
3 3 3 Blood
4 7 7 Blood
5 11 14 Blood
6 32 30 Blood
visit
1 1
2 2
3 3
4 4
5 5
6 6abdata <- inner_join(ab_titer, meta)Joining with `by = join_by(specimen_id)`head(abdata) specimen_id isotype is_antigen_specific antigen MFI MFI_normalised
1 1 IgE FALSE Total 1110.21154 2.493425
2 1 IgE FALSE Total 2708.91616 2.493425
3 1 IgG TRUE PT 68.56614 3.736992
4 1 IgG TRUE PRN 332.12718 2.602350
5 1 IgG TRUE FHA 1887.12263 34.050956
6 1 IgE TRUE ACT 0.10000 1.000000
unit lower_limit_of_detection subject_id infancy_vac biological_sex
1 UG/ML 2.096133 1 wP Female
2 IU/ML 29.170000 1 wP Female
3 IU/ML 0.530000 1 wP Female
4 IU/ML 6.205949 1 wP Female
5 IU/ML 4.679535 1 wP Female
6 IU/ML 2.816431 1 wP Female
ethnicity race year_of_birth date_of_boost dataset
1 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
2 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
3 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
4 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
5 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
6 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
actual_day_relative_to_boost planned_day_relative_to_boost specimen_type
1 -3 0 Blood
2 -3 0 Blood
3 -3 0 Blood
4 -3 0 Blood
5 -3 0 Blood
6 -3 0 Blood
visit
1 1
2 1
3 1
4 1
5 1
6 1Q. What Antibody isotypes are measured for these patients?
table(abdata$isotype)
IgE IgG IgG1 IgG2 IgG3 IgG4
6698 7265 11993 12000 12000 12000 Q. What antigens are reported?
table(abdata$antigen)
ACT BETV1 DT FELD1 FHA FIM2/3 LOLP1 LOS Measles OVA
1970 1970 6318 1970 6712 6318 1970 1970 1970 6318
PD1 PRN PT PTM Total TT
1970 6712 6712 1970 788 6318 Let’s focus on the IgG antigen and make a plot of MFI_normalized for all anitgens.
igg <- abdata |>
filter(isotype == "IgG")
head(igg) specimen_id isotype is_antigen_specific antigen MFI MFI_normalised
1 1 IgG TRUE PT 68.56614 3.736992
2 1 IgG TRUE PRN 332.12718 2.602350
3 1 IgG TRUE FHA 1887.12263 34.050956
4 19 IgG TRUE PT 20.11607 1.096366
5 19 IgG TRUE PRN 976.67419 7.652635
6 19 IgG TRUE FHA 60.76626 1.096457
unit lower_limit_of_detection subject_id infancy_vac biological_sex
1 IU/ML 0.530000 1 wP Female
2 IU/ML 6.205949 1 wP Female
3 IU/ML 4.679535 1 wP Female
4 IU/ML 0.530000 3 wP Female
5 IU/ML 6.205949 3 wP Female
6 IU/ML 4.679535 3 wP Female
ethnicity race year_of_birth date_of_boost dataset
1 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
2 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
3 Not Hispanic or Latino White 1986-01-01 2016-09-12 2020_dataset
4 Unknown White 1983-01-01 2016-10-10 2020_dataset
5 Unknown White 1983-01-01 2016-10-10 2020_dataset
6 Unknown White 1983-01-01 2016-10-10 2020_dataset
actual_day_relative_to_boost planned_day_relative_to_boost specimen_type
1 -3 0 Blood
2 -3 0 Blood
3 -3 0 Blood
4 -3 0 Blood
5 -3 0 Blood
6 -3 0 Blood
visit
1 1
2 1
3 1
4 1
5 1
6 1ggplot(igg) +
aes(MFI_normalised, antigen) +
geom_boxplot()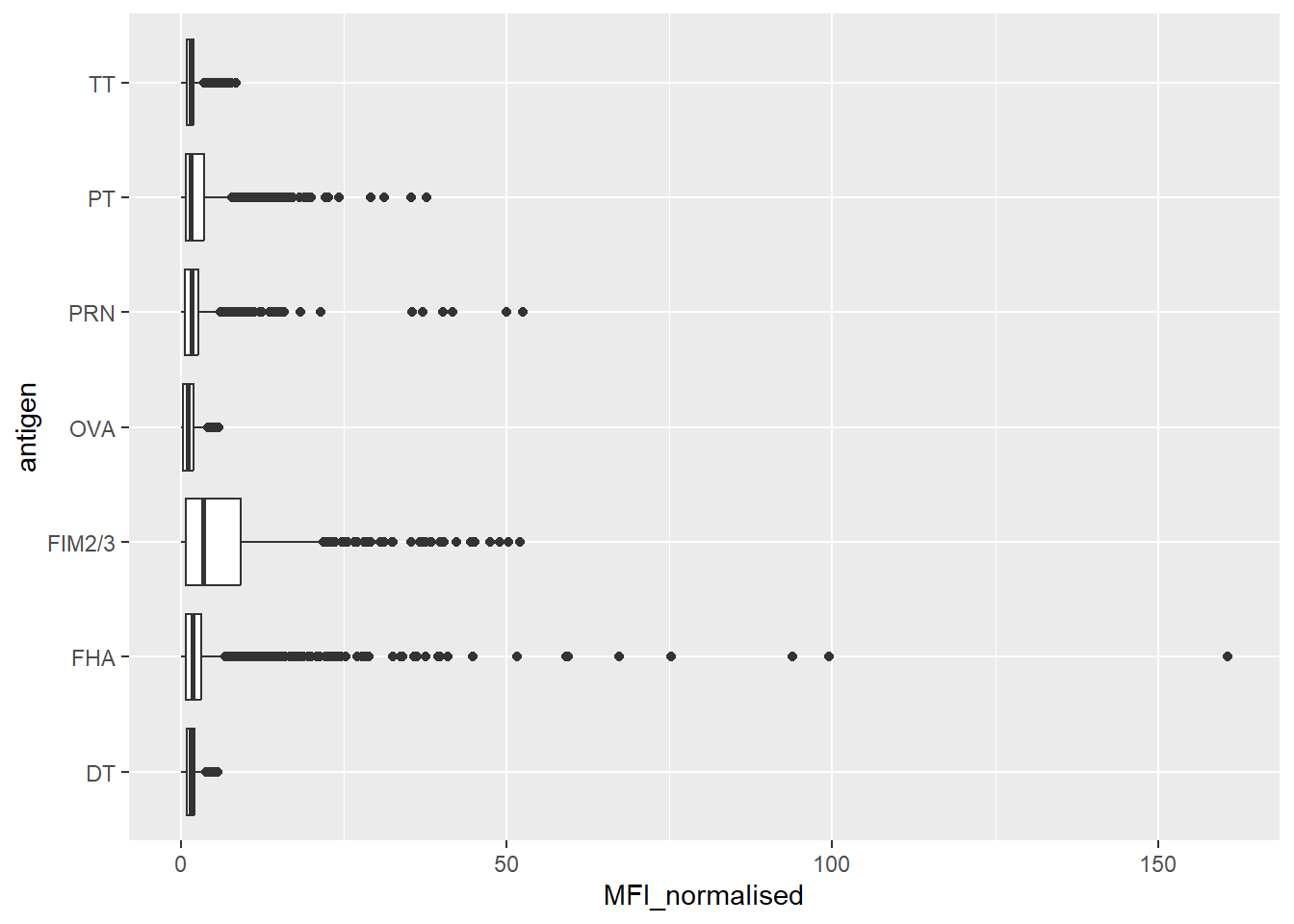
Q. Is there a difference for aP vs wP individuals with these values?
ggplot(igg) +
aes(MFI_normalised, antigen) +
geom_boxplot() +
facet_wrap(~infancy_vac)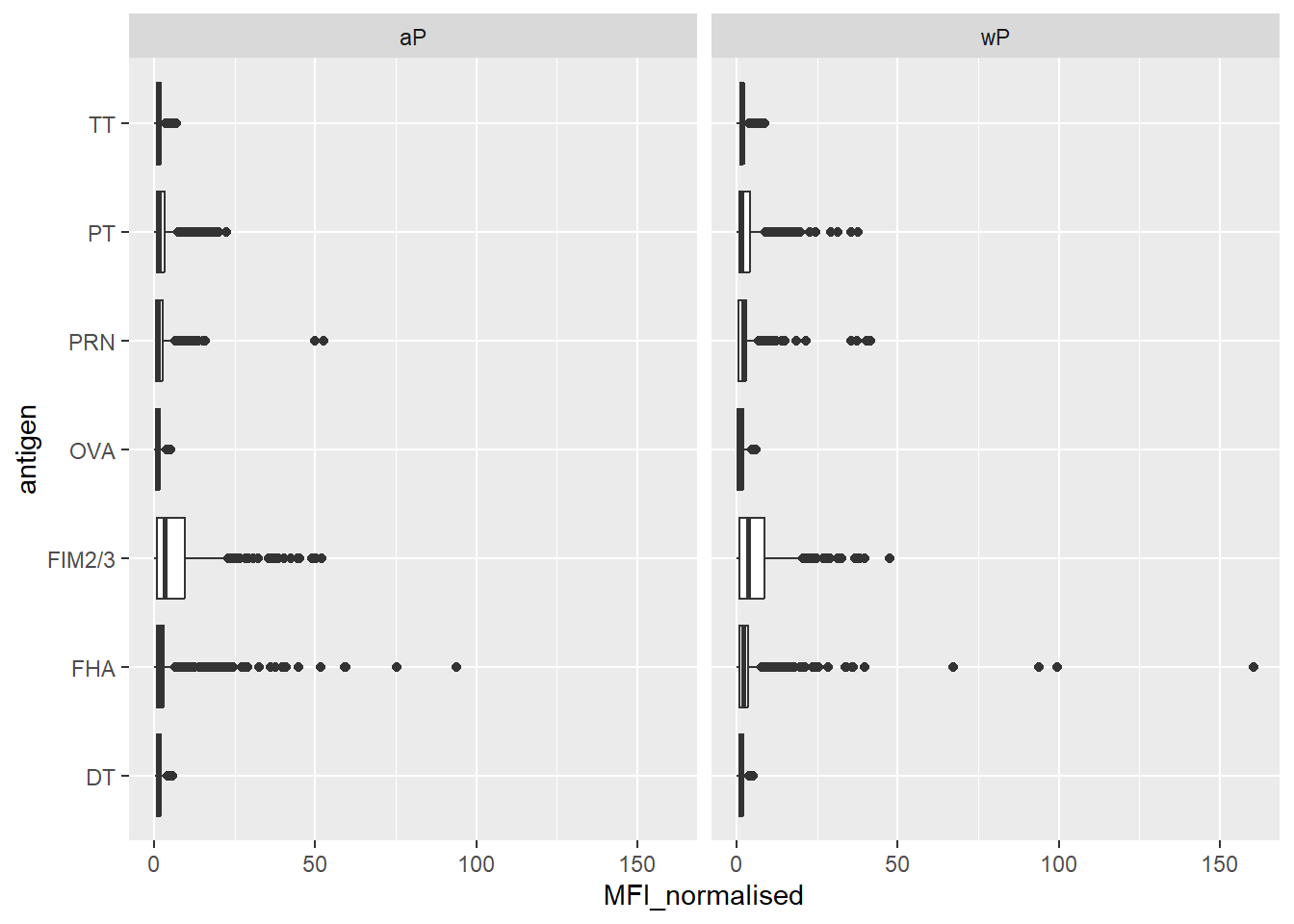
ggplot(igg) +
aes(MFI_normalised, antigen, col=infancy_vac) +
geom_boxplot()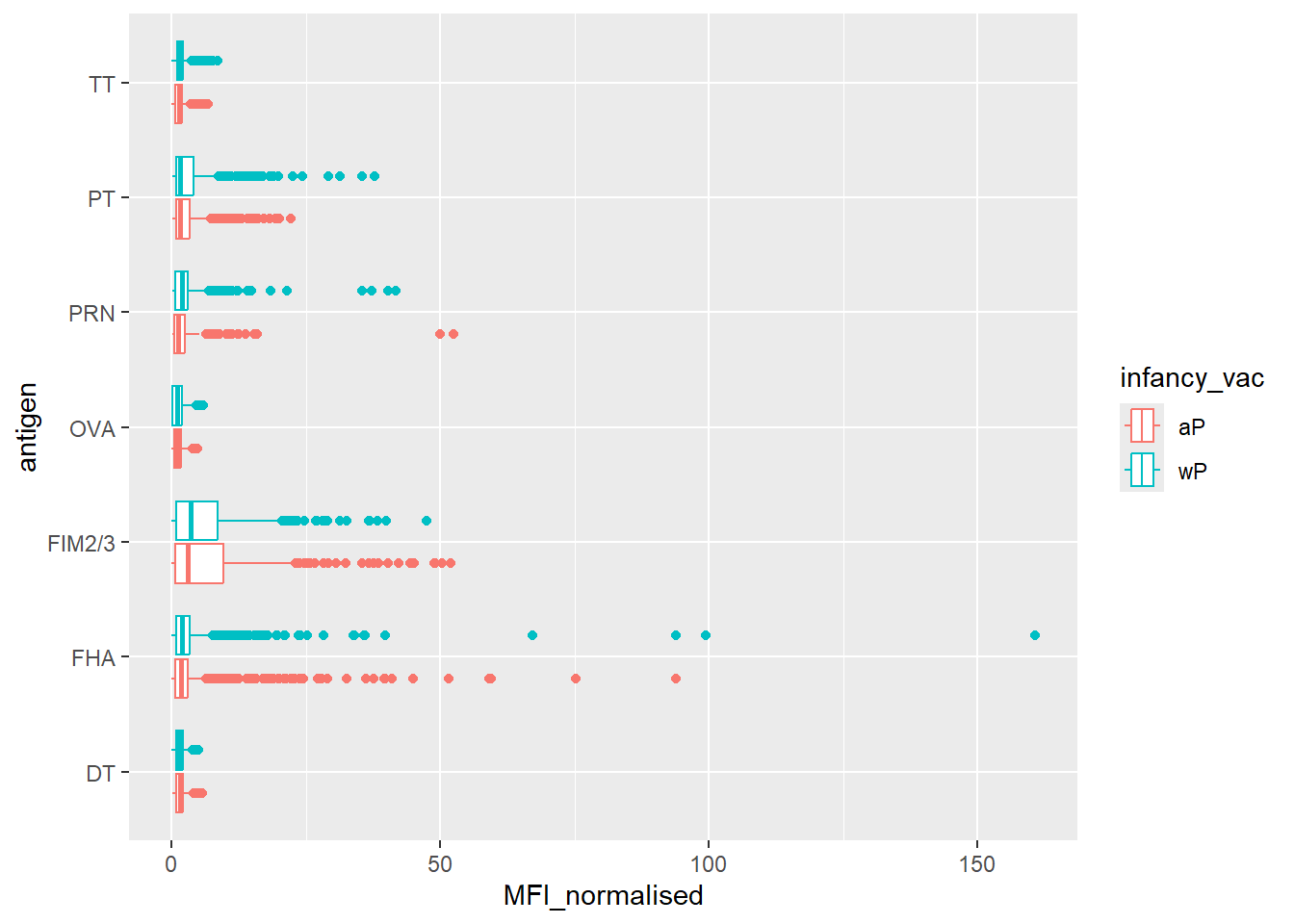
Q. Is there a temprol response - i.e do valujes increase or decrease over time?
ggplot(igg) +
aes(MFI_normalised, antigen, col=infancy_vac) +
geom_boxplot() +
facet_wrap(~visit)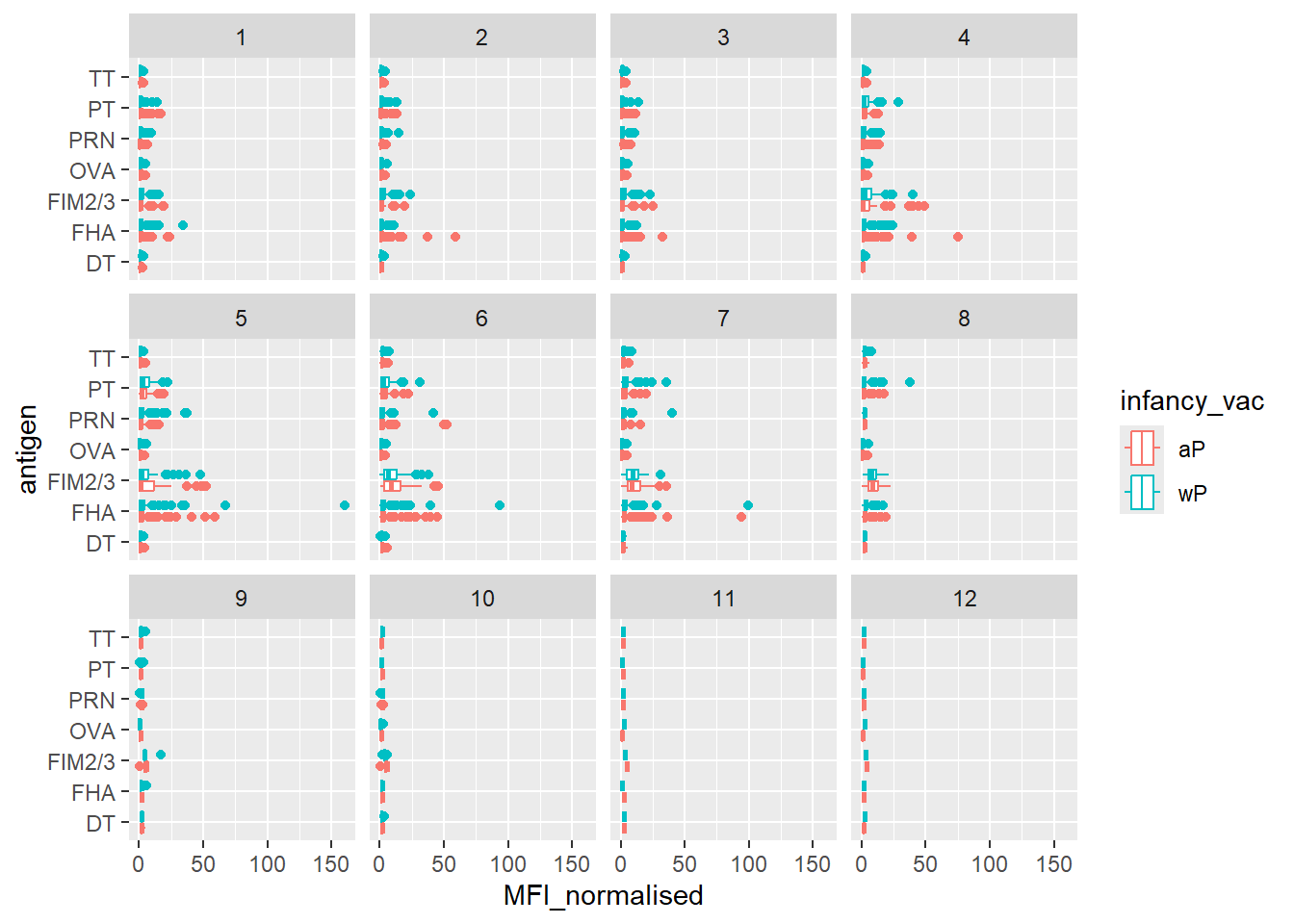
Focus on “PT” Pertusisis toxin antigen
pt.igg.21 <- igg |> filter(antigen == "PT",
dataset== "2021_dataset")ggplot(pt.igg.21) +
aes(planned_day_relative_to_boost, MFI_normalised, col=infancy_vac, group = subject_id) +
geom_point() +
geom_line() +
geom_vline(xintercept = 14, lty=2)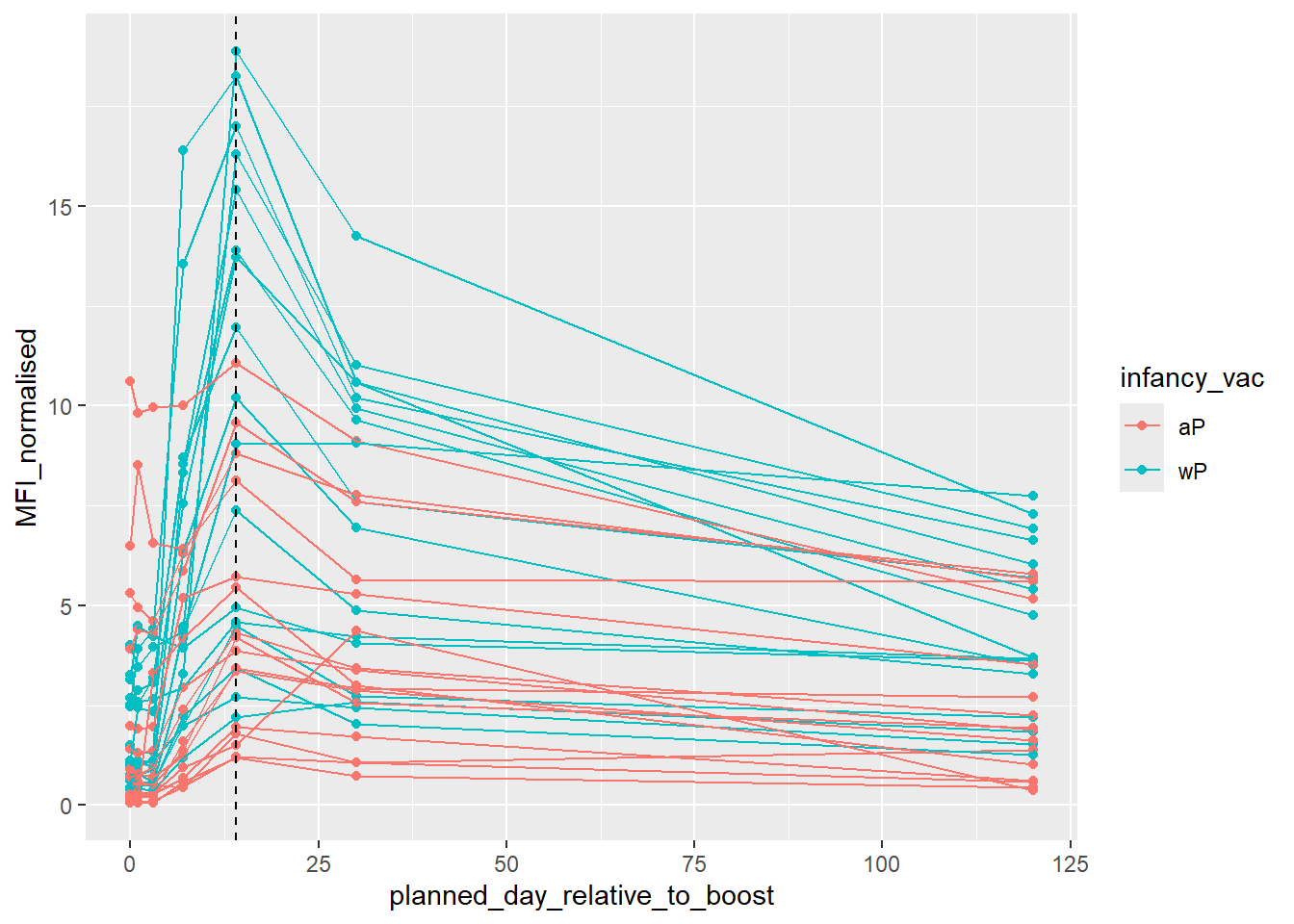
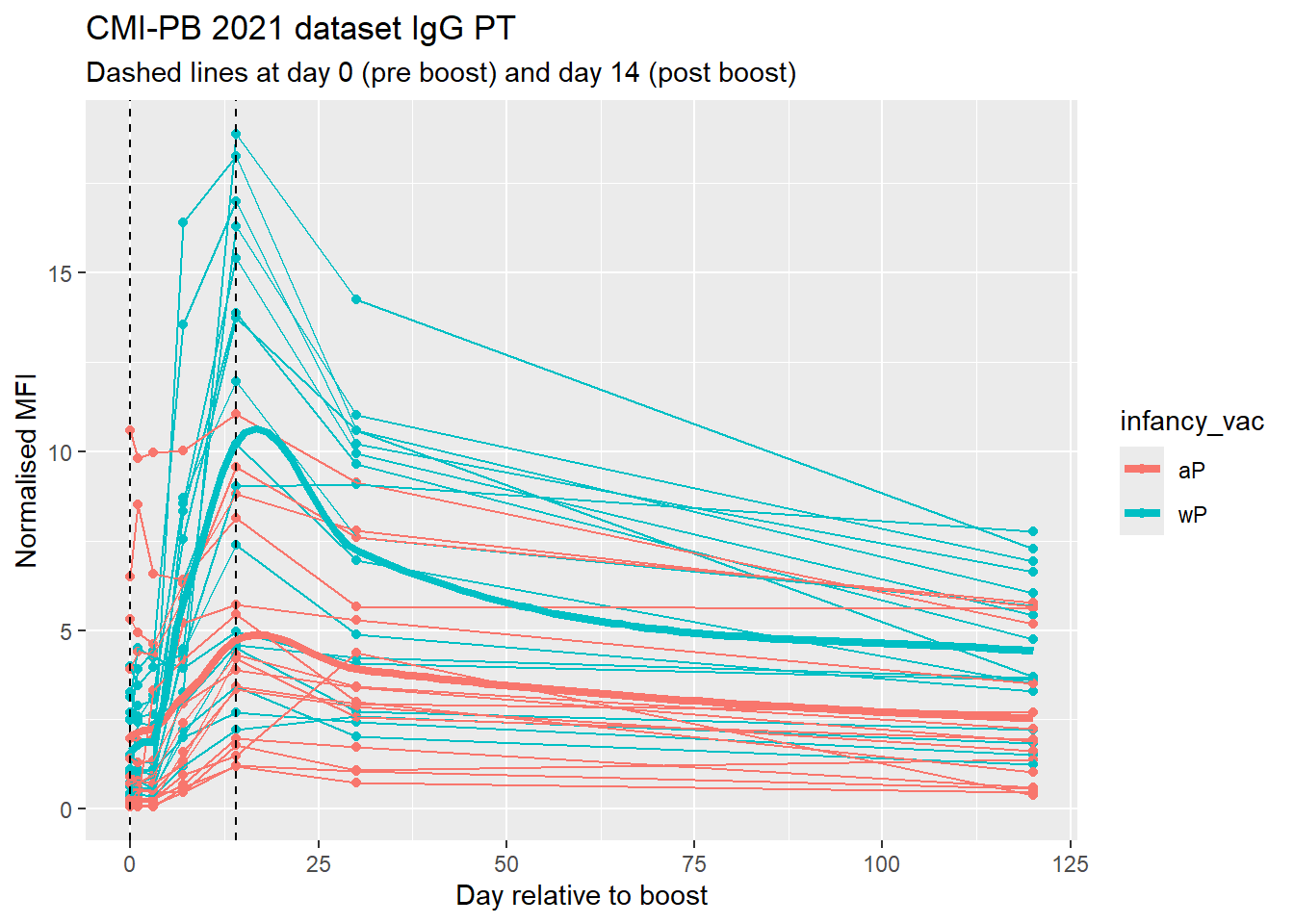
Final Graph
ggplot(pt.igg.21) +
aes(planned_day_relative_to_boost, MFI_normalised, col=infancy_vac, group = subject_id) +
geom_point() +
geom_line() +
geom_vline(xintercept = 14, lty=2) +
geom_vline(xintercept = 0, lty=2) +
labs(
title = "CMI-PB 2021 dataset IgG PT",
subtitle = "Dashed lines at day 0 (pre boost) and day 14 (post boost)",
x = "Day relative to boost",
y = "Normalised MFI"
) +
geom_smooth(aes(x = planned_day_relative_to_boost, y = MFI_normalised, group = infancy_vac), se= FALSE, , size = 1.5, span = .3)Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: pseudoinverse used at -0.6Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: neighborhood radius 3.6Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: reciprocal condition number 0Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: There are other near singularities as well. 11364Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: pseudoinverse used at -0.6Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: neighborhood radius 3.6Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: reciprocal condition number 2.4057e-16Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: There are other near singularities as well. 11364