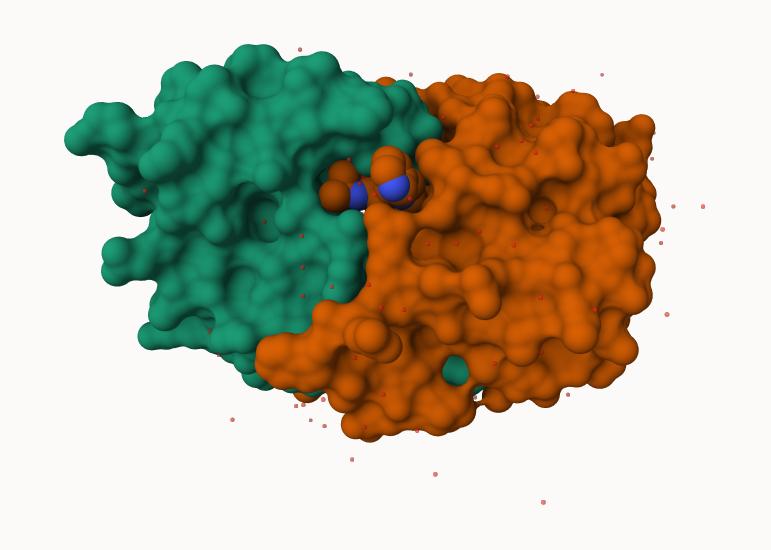
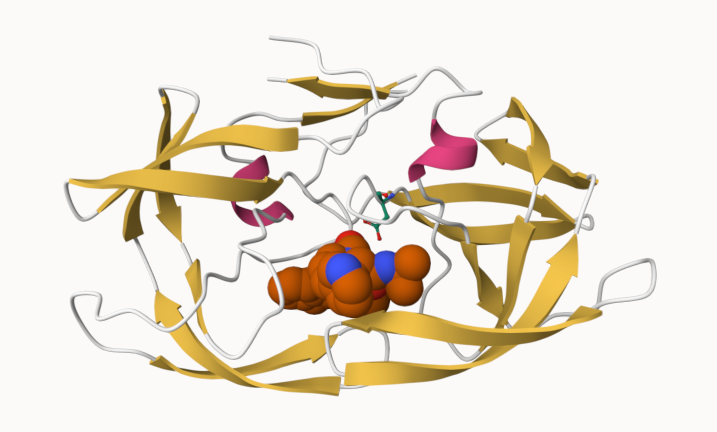
Q. Generate and insert an image of the HIV-Pr cartoon colored by secondary structure, showing the inhibitor (ligand) in spacefill.
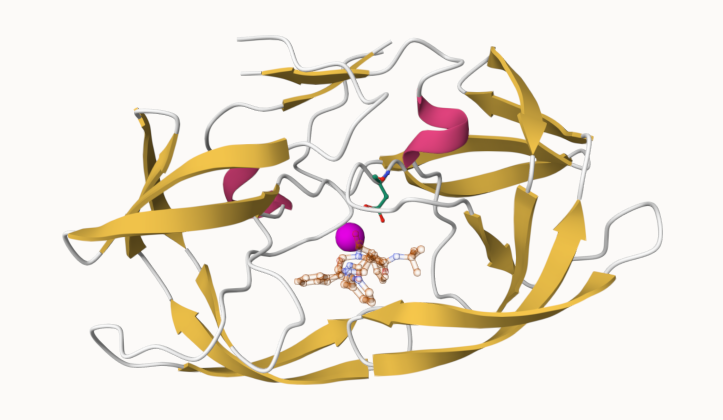
Q. One fimal image showing catalytic APS 25 and the all-important active site water molecule.
The bio3D package for structural bioinformatics
library(bio3d)hiv <-read.pdb("1hsg")
Note: Accessing on-line PDB file
hiv
Call: read.pdb(file = "1hsg")
Total Models#: 1
Total Atoms#: 1686, XYZs#: 5058 Chains#: 2 (values: A B)
Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
Non-protein/nucleic Atoms#: 172 (residues: 128)
Non-protein/nucleic resid values: [ HOH (127), MK1 (1) ]
Protein sequence:
PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
VNIIGRNLLTQIGCTLNF
+ attr: atom, xyz, seqres, helix, sheet,
calpha, remark, call
head( hiv$atom )
type eleno elety alt resid chain resno insert x y z o b
1 ATOM 1 N <NA> PRO A 1 <NA> 29.361 39.686 5.862 1 38.10
2 ATOM 2 CA <NA> PRO A 1 <NA> 30.307 38.663 5.319 1 40.62
3 ATOM 3 C <NA> PRO A 1 <NA> 29.760 38.071 4.022 1 42.64
4 ATOM 4 O <NA> PRO A 1 <NA> 28.600 38.302 3.676 1 43.40
5 ATOM 5 CB <NA> PRO A 1 <NA> 30.508 37.541 6.342 1 37.87
6 ATOM 6 CG <NA> PRO A 1 <NA> 29.296 37.591 7.162 1 38.40
segid elesy charge
1 <NA> N <NA>
2 <NA> C <NA>
3 <NA> C <NA>
4 <NA> O <NA>
5 <NA> C <NA>
6 <NA> C <NA>